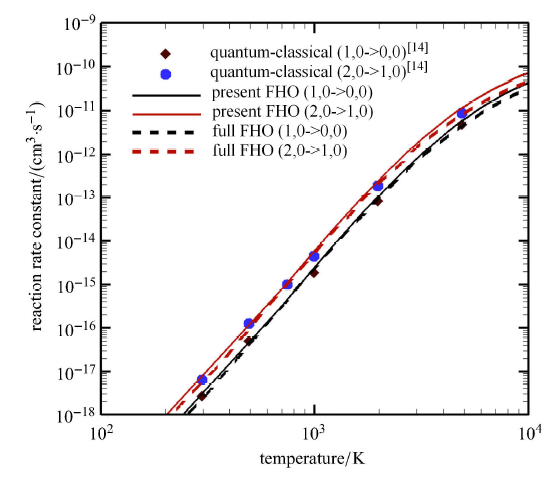
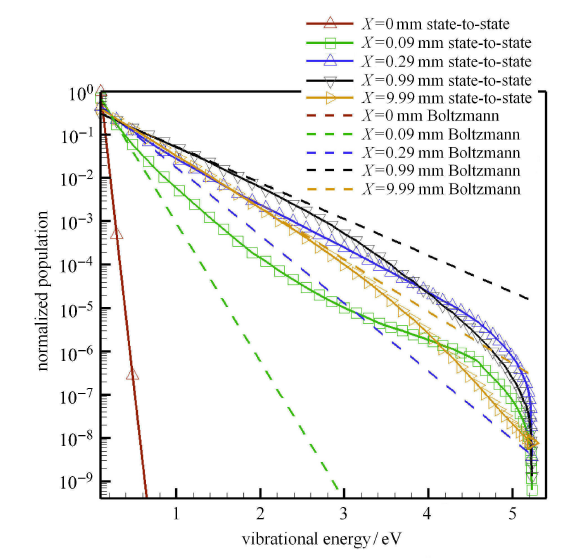
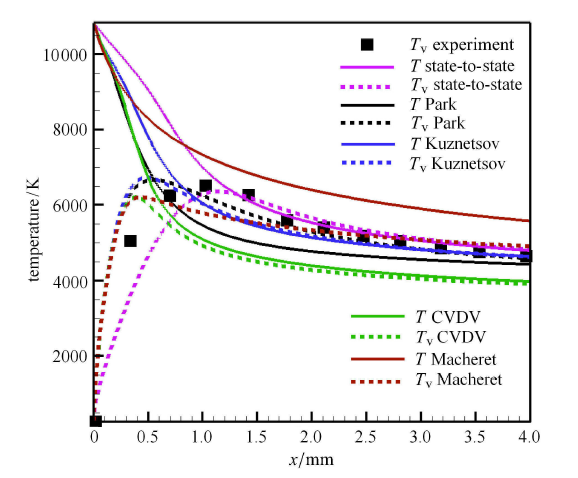
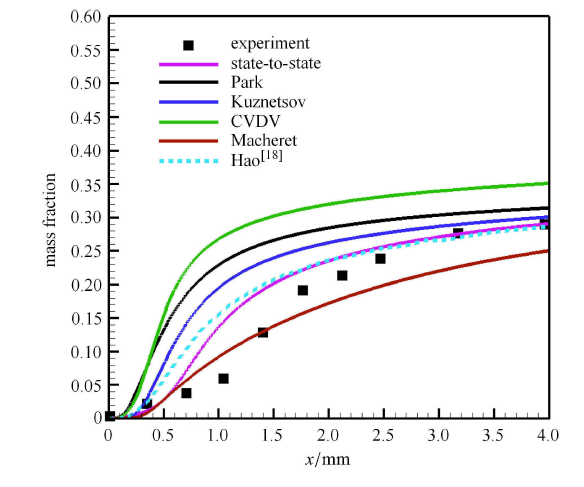
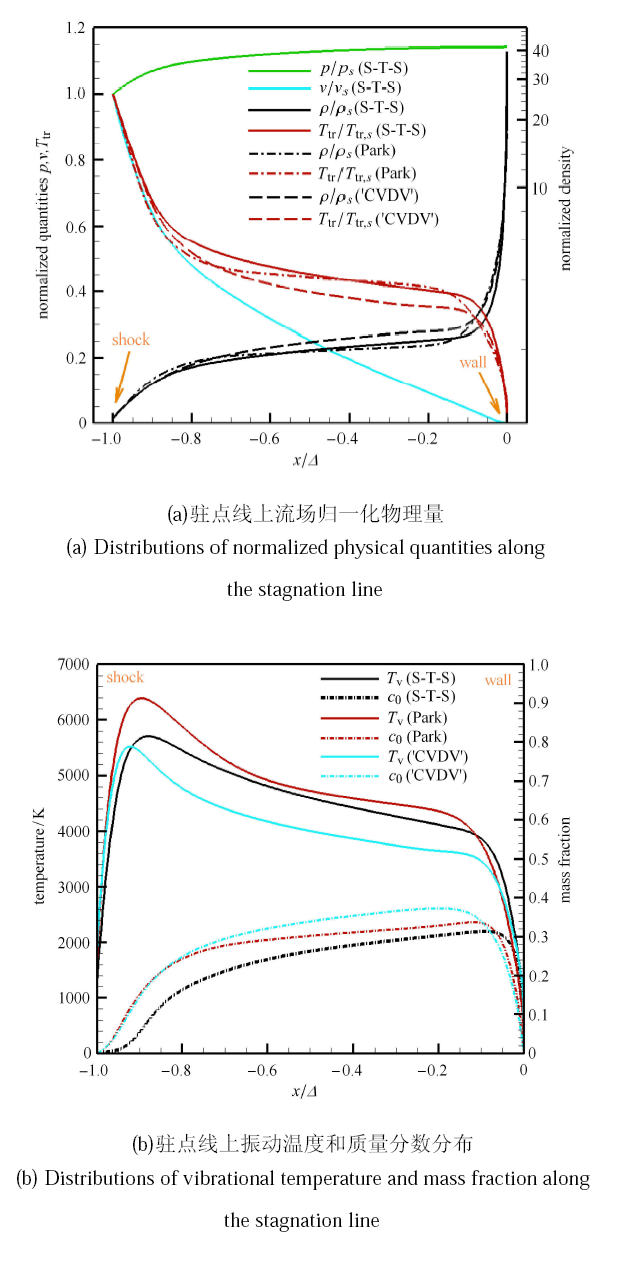
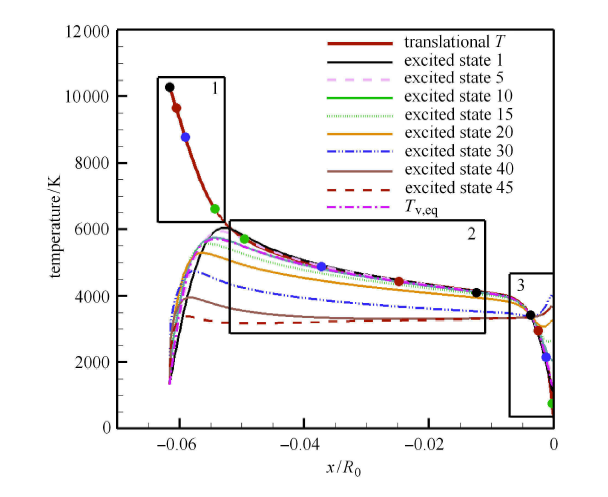
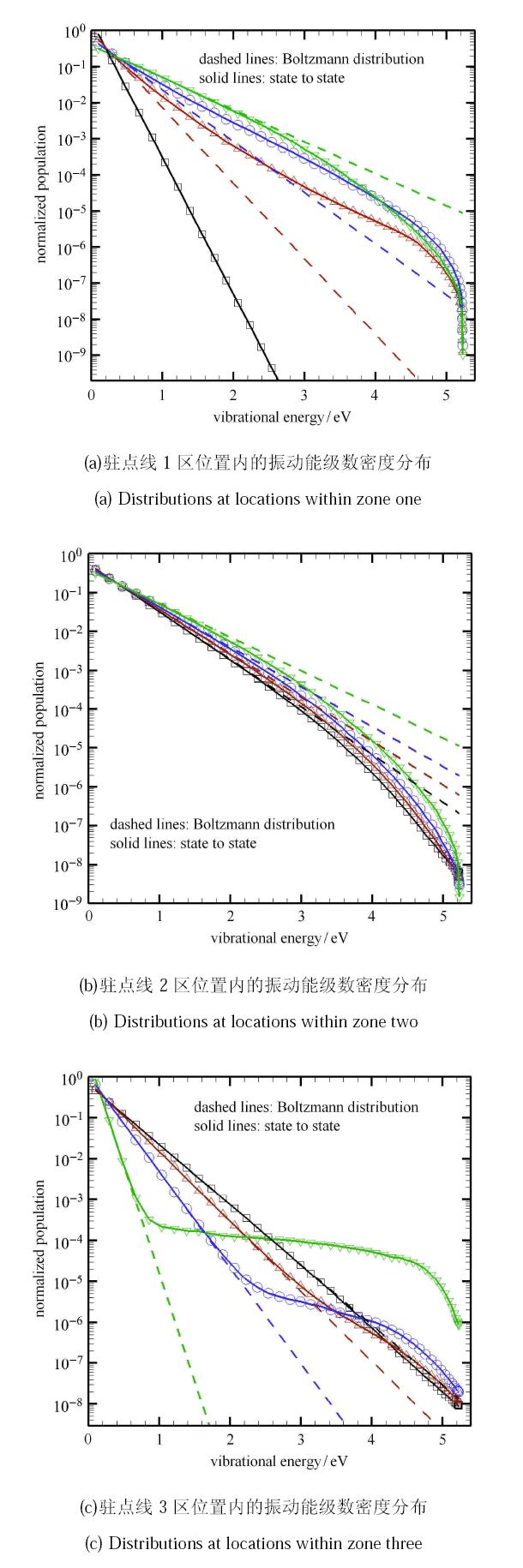
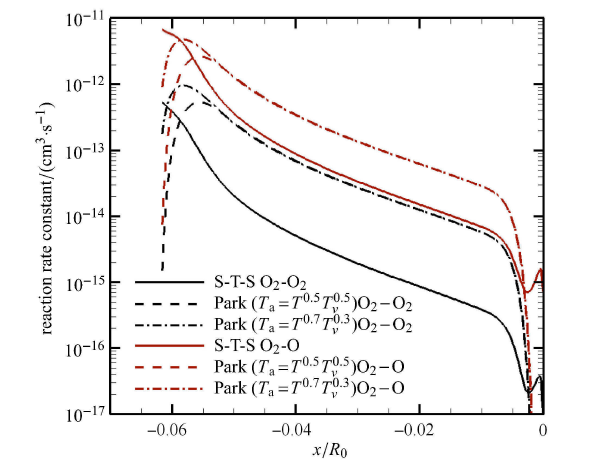
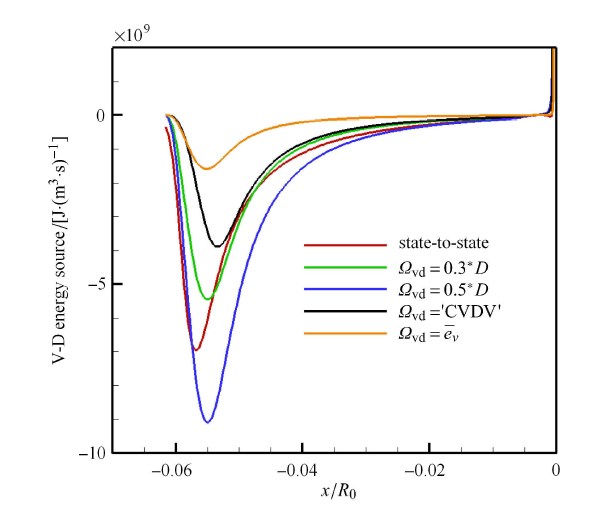
Hypersonic flow is usually in a thermochemical nonequilibrium state due to high temperature after the bow shock. In this paper, the state-to-state method and two-temperature models are employed to study the thermochemical nonequilibrium processes of oxygen for a post-shock flow and a flow over a blunt body along the stagnation line. The state-to-state method treats each vibrational energy level of molecular oxygen as an independent species, and predicts the number density of each vibrational level by coupling the Euler equations or reduced Navier-Stokes equations along the stagnation line. The two-temperature models assume that all vibrational levels follow the Boltzmann distribution at a vibrational temperature, and a vibrational energy equation is solved to obtain the vibrational temperature. Simulation results show that the distributions of the temperature and species concentration predicted by the state-to-state method are in good agreement with the available experimental results in the literature, while the classical two-temperature models show large errors and the results of different two-temperature models are scattered. The state-to-state method gives detailed information of all vibrational levels along the streamline. After the normal shock or bow shock, the high vibrational levels are first excited but low levels with large number density will reach thermal equilibrium first, whereas high level molecules reach thermal equilibrium only after a long distance. Near the stagnation point, the recombination reaction produces oxygen molecules that are at high vibrational levels, thus the number density of a high vibration level is significantly higher than that of the equilibrium distribution. It is also found that the dissociation rate of classical two-temperature models deviates from the state-to-state result, which cannot accurately account for the coupling effects of vibration dissociation on the dissociation rate. However, it is reasonable for Park’s two-temperature model to take the vibration energy lost by dissociation to be 0.3$\sim$0.5 times of the molecular dissociation energy.
Keywords:high temperature non-equilibrium
;
vibrational relaxation
;
state-to-state calculation
;
stagnation line model
Hong Qizhen, Wang Xiaoyong, Sun Quanhua. DETAILED ANALYSIS OF VIBRATIONAL STATES OF OXYGEN IN HIGH TEMPERATURE NON-EQUILIBRIUM FLOWS1). Chinese Journal of Theoretical and Applied Mechanics[J], 2019, 51(6): 1761-1774 DOI:10.6052/0459-1879-19-145
A hypersonic vehicle traveling at a high speed disrupts the distribution of internal states in the ambient flow and introduces a nonequilibrium distribution in the post-shock conditions. We investigate the vibrational relaxation in diatom-atom collisions in the range of temperatures between 1000 and 10 000 K by comparing results of extensive fully quantum-mechanical and quasi-classical simulations with available experimental data. The present paper simulates the interaction of molecular oxygen with argon as the first step in developing the aerothermodynamics models based on first principles. We devise a routine to standardize such calculations also for other scattering systems. Our results demonstrate very good agreement of vibrational relaxation time, derived from quantum-mechanical calculations with the experimental measurements conducted in shock tube facilities. At the same time, the quasi-classical simulations fail to accurately predict rates of vibrationally inelastic transitions at temperatures lower than 3000 K. This observation and the computational cost of adopted methods suggest that the next generation of high fidelity thermochemical models should be a combination of quantum and quasi-classical approaches.
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.
Ultrasound is now a clinically-accepted modality in the management of osteoporosis. The most common commercial clinical devices assess fracture risk from measurements of attenuation and sound speed in cancellous bone. This review discusses fundamental mechanisms underlying the interaction between ultrasound and cancellous bone. Because of its two-phase structure (mineralized trabecular network embedded in soft tissue-marrow), its anisotropy, and its inhomogeneity, cancellous bone is more difficult to characterize than most soft tissues. Experimental data for the dependences of attenuation, sound speed, dispersion, and scattering on ultrasound frequency, bone mineral density, composition, microstructure, and mechanical properties are presented. The relative roles of absorption, scattering, and phase cancellation in determining attenuation measurements in vitro and in vivo are delineated. Common speed of sound metrics, which entail measurements of transit times of pulse leading edges (to avoid multipath interference), are greatly influenced by attenuation, dispersion, and system properties including center frequency and bandwidth. However, a theoretical model has been shown to be effective for correction for these confounding factors in vitro and in vivo. Theoretical and phantom models are presented to elucidate why cancellous bone exhibits negative dispersion, unlike soft tissue, which exhibits positive dispersion. Signal processing methods are presented for separating "fast" and "slow" waves (predicted by poro-elasticity theory and supported in cancellous bone) even when the two waves overlap in time and frequency domains. Models to explain dependences of scattering on frequency and mean trabecular thickness are presented and compared with measurements. Anisotropy, the effect of the fluid filler medium (marrow in vivo or water in vitro), phantoms, computational modeling of ultrasound propagation, acoustic microscopy, and nonlinear properties in cancellous bone are also discussed.
MillikanRC, WhiteDR.
Systematics of vibrational relaxation
Journal of Chemical Physics, 1963,39(12):3209-3213
The picosecond optical-optical double resonance experiment in a supersonic free jet as well as the vapor-phase phosphorescence indicates that the decay of T(1) Cl(2)CS belongs to the intermediate case of the classification scheme for electronic relaxation. The A(fast)/A(slow) pre-exponential ratio in the biexponential T(1) decay is much greater under picosecond excitation than under nanosecond excitation. In vapor phase at low pressure, the phosphorescence exhibits a decay time that varies with the coherence width of the laser used for excitation. Both the T(1) and the S(1) decay times of Cl(2)CS depend strongly on temperature, indicating that Coriolis coupling plays an important role in mode mixing (intramolecular vibrational redistribution).
SchwartzRN, SlawskyZI, HerzfeldKF.
Calculation of vibrational relaxation times in gases
Journal of Chemical Physics, 1952,20(10):1591-1599
This work presents a theoretical treatment of the vibrational line shape generated in a femtosecond stimulated Raman spectroscopy (FSRS) experiment under conditions in which the probed vibration undergoes a significant frequency shift during its free induction decay. This theory is applied to simulate the FSRS lineshapes previously observed in rhodopsin (Kukura et al. Science 2005, 310, 1006). The previously determined relaxation times for formation of the trans-photoproduct of rhodopsin were calculated using an incorrect equation for the time dependence of the observed frequency shifts. Here the data are reanalyzed by calculation of the corrected frequency sweep occurring during the vibrational free induction decay. It is shown that the calculated frequency shifts and general conclusions of the original work are sound but that the coherent vibrational frequency shifts of the C(10), C(11), and C(12) hydrogen-out-of-plane vibrations occur with a 140 fs time constant rather than the previously reported 325 fs time constant. This time constant provides an important constraint for models of the dynamics of the cis to trans isomerization process.
KernerEH.
Note on the forced and damped oscillator in quantum mechanics
In this work, a computational model of state-to-state energy flow in gas ensembles is used to investigate collisional relaxation of excited OH, present as a minor species in various bath gases. Rovibrational quantum state populations are computed for each component species in ensembles consisting of 8000 molecules undergoing cycles of binary collisions. Results are presented as quantum state populations and as (approximate) modal temperatures for each species after each collision cycle. Equilibration of OH is slow with Ar as the partner but much faster when N(2) and/or O(2) forms the bath gas. This accelerated thermalization is shown to be the result of near-resonant vibration-vibration transfer, with vibrational de-excitation in OH matched in energy by excitation in bath molecules. Successive near-resonant events result in an energy cascade. Such processes are highly dependent on molecule pair and on initial OH vibrational state. OH rotational temperatures initially increase, but at equilibration, they are lower than those of other modes. Possible reasons for this observation in molecules such as OH are suggested. There are indications of an order of precedent in the equilibration process, with vibrations taking priority over rotations, and potential explanations for this phenomenon are discussed.
BillingGD, KolesnickRE.
Vibrational relaxation of oxygen: state to state rate constants
The oxygen absorbance was studied at wavelengths 200-270 nm in Schumann-Runge system behind the front of a strong shock wave. Using these data, the vibrational temperature Tv behind the front of shock waves was measured at temperatures 4000-10,800 K in undiluted oxygen. Determination of Tv was based on the measurements of time histories of absorbance for two wavelengths behind the shock front and on the results of detail calculations of oxygen absorption spectrum. Solving the system of standard quasi-one-dimensional gas dynamics equations and using the measured vibrational temperature, the time evolution of oxygen concentration and other gas parameters in each experiment were calculated. Based on these data, the oxygen dissociation rate constants were obtained for thermal equilibrium and thermal non-equilibrium conditions. Furthermore, the oxygen vibrational relaxation time was also determined at high temperatures. Using the experimental data, various theoretical and empirical models of high-temperature dissociation were tested, including the empirical model proposed in the present work.
ColettiC, BillingGD.
Vibrational energy transfer in molecular oxygen collisions
Vibrational relaxation of O2(X 3sigma(g)-, upsilon=2,3) by O2 molecules is studied via a two-laser approach. Laser radiation at 266 nm photodissociates ozone in a mixture of molecular oxygen and ozone. The photolysis step produces vibrationally excited O2(a 1delta(g)) that is rapidly converted to O2(X 3sigma(g)-, upsilon=2,3) in a near-resonant adiabatic electronic energy-transfer process involving collisions with ground-state O2. The output of a tunable 193-nm ArF laser monitors the temporal evolution of the O2(X 3sigma(g)-, upsilon=2,3) population via laser-induced fluorescence detected near 360 nm. The rate coefficients for the vibrational relaxation of O2(X 3sigma(g)-, upsilon=2,3) in collision with O2 are 2.0(-0.4)(+0.6) x 10(-13) cm3 s(-1) and (2.6+/-0.4) x 10(-13) cm3 s(-1), respectively. These rate coefficients agree well with other experimental work but are significantly larger than those produced by various semiclassical theoretical calculations.
The oxygen absorbance was studied at wavelengths 200-270 nm in Schumann-Runge system behind the front of a strong shock wave. Using these data, the vibrational temperature Tv behind the front of shock waves was measured at temperatures 4000-10,800 K in undiluted oxygen. Determination of Tv was based on the measurements of time histories of absorbance for two wavelengths behind the shock front and on the results of detail calculations of oxygen absorption spectrum. Solving the system of standard quasi-one-dimensional gas dynamics equations and using the measured vibrational temperature, the time evolution of oxygen concentration and other gas parameters in each experiment were calculated. Based on these data, the oxygen dissociation rate constants were obtained for thermal equilibrium and thermal non-equilibrium conditions. Furthermore, the oxygen vibrational relaxation time was also determined at high temperatures. Using the experimental data, various theoretical and empirical models of high-temperature dissociation were tested, including the empirical model proposed in the present work.
ParkC.
Assessment of two-temperature kinetic model for ionizing air
Journal of Thermophysics & Heat Transfer, 1987,3(3):233-244
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.
ChernyiG, LosevS, MacheretS, et al.
Physical and chemical processes in gas dynamics: cross sections and rate constants, Volume I
Progress in Astronautics and Aeronautics, 2002,196
MarroneP, TreanorC.
Chemical relaxation with preferential dissociation from excited vibrational levels
The polarization singularities are directly generated by using plasmonic metasurfaces with the geometric phase profiles designed to form the Poincaré beams. Different morphologies of polarization topological structures of lemon, star, monstar, spiral, dipole and quadrupole are created by the superpositions of Laguerre-Gauss modes with different orders under orthogonal circular or linear polarization basis. The polarization ellipse patterns and topological features of the produced optical vector fields are analyzed to reveal the properties of the polarization singularities of C-points and L-lines, and the orbital angular momentum states are also measured. The demonstrated polarization singularities generated from the geometric metasurfaces will promise many potential applications related to optical polarization imaging, metrology, optical trapping and quantum information processing.
HammerlingP, TeareJD, KivelB.
Theory of radiation from luminous shock waves in nitrogen
The spectroscopic Franck-Condon (FC) principle is extended to mechanochemistry. If the external force is applied rapidly (the sudden-force regime), then the transition between the potential energy surface and the force-modified potential energy surface is analogous to the optical electronic transition. Such a transition produces a nonequilibrium ensemble of vibrationally excited molecules. This excess of vibrational energy is another activation source in addition to the well-known reaction barrier modulation by the external force. In the same time, the nonequilibrium vibrational distribution implies nonstatistical kinetics of a mechanochemical transformation. Mechanochemical FC principle thus provides a conceptual picture for the sudden-force mechanochemistry and opens possibilities for quantitative calculations of the mechanochemical rates and mechanisms. Here we use it to compute the dissociation rates of a model diatomic molecule and to explain the selectivity in mechanochemical bond breaking in n-butane. The approach is predicted to be relevant for large-magnitude external forces, applied instantaneously. Otherwise, the excess vibrational energy will dissipate due to intramolecular vibrational redistribution and interaction with environment.
In the case of nuclear incidents, radioiodine may be liberated. After incorporation it accumulates in the thyroid and by internal irradiation enhances the risk of cancer occurrence. By administering a large dose of non-radioactive iodine the uptake of radioiodine into the gland can be inhibited ("iodine blockade"). Biokinetic models using first order kinetics are not suited to simulate iodine blockade, as the uptake into the gland is mediated by a saturable active transport. Therefore, we integrated an uptake mechanism described by a Michaelis-Menten kinetic into a simple ICRP biokinetic model. We moreover added a total uptake blocking mechanism representing the Wolff-Chaikoff effect becoming active when the gland is saturated with iodine. The validity of the model was ascertained by comparison with IMBA software. The competition of radioiodine and stable iodine at the membrane carrier site was modeled according to the rate law for monomolecular reactions for competing substrates. Our simulations show that competition for the uptake at the membrane carrier site accounts for about 60% and the saturation of the gland with iodine for over 35% of the total protective efficacy that exceeds 95%. Following acute radioiodine exposure, it is preferable to administer a single large dose of stable iodine. In the case of continuous radioiodine exposure, a single dose of stable iodine is less effective than after an acute exposure and splitting the total available dose and shortening the dosage intervals enhance efficacy. Model-based simulations may be a useful tool to develop antidote dosage schemes for uncommon emergencies.
AdamovichIV, MacheretSO, RichJW, et al.
Vibrational energy transfer rates using a forced harmonic oscillator model
Journal of Thermophysics & Heat Transfer, 1998,12(1):57-65
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.
AndrienkoDA, BoydID.
High fidelity modeling of thermal relaxation and dissociation of oxygen
A review of reaction rates and thermodynamic and transport properties for the 11-species air model for chemical and thermal nonequilibrium calculations to 30 000 K
NASA Technical Report, 1989,89(6):32-34
KustovaEV.
On the simplified state-to-state transport coefficients
The near-space solar-powered unmanned aerial vehicle has broad prospects in application owing to its high altitude long-endurance performance. Launching solar-powered unmanned aerial vehicle into the near-space with balloon-borne approach has advantages over the traditional sliding take-off methods, in that it is able to quickly and safely cross the turbulent zone. In this article, we investigate the control technology of balloon-borne launching for the solar-powered unmanned aerial vehicles. First, the motion of the launching process is divided into longitudinal and lateral-directional motion, with the longitudinal process and its equation addressed in detail. We then analyze the flight state and restriction conditions that the unmanned aerial vehicle should meet during the process. Second, the target variables and constraints are selected to formulate the optimization problem. The control variable parameterization method is applied to find the optimal pitch angle in the releasing-and-pulling process. More explicitly, a three-channel attitude stabilization controller is designed, in which the longitudinal channel takes the optimal pitch angle as the pitch instruction, the transverse channel carries out the zero control of the inclination angle, and the course channel takes the stabilization control, respectively. Numerical simulation results show that our proposed control design is capable of accelerating the solar-powered unmanned aerial vehicles from the vertical state and pulling them up to the horizontal cruising flight state, with the flight angle of attack, the maximum speed, and the maximum axial acceleration in the pulling process all within the designed range.
A review of reaction rates and thermodynamic and transport properties for the 11-species air model for chemical and thermal nonequilibrium calculations to 30 000 K
1989
On the simplified state-to-state transport coefficients






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}