The non-equilibrium phenomenon of thermochemical coupling has been a difficult problem in high temperature aerothermal dynamics, and hinders to analyze phenomena such as cell structure of detonation wave and ignition speed of low temperature combustion. In this paper, typical chemical reaction models (TCE, VFD, QK models) employed in the direct simulation Monte Carlo (DSMC) simulation are analyzed using two examples (namely, N dissociation at high temperature, and chain displacement reaction in H‒ O combustion) from microscopic reaction probability, vibrational state specific reaction rates, total reaction rate under thermal nonequilibrium condition, and post-collision redistribution of internal energy. It is found that the probability distribution of vibrational energy of reacted molecules deviates from the equilibrium Boltzmann distribution for both the high temperature dissociation reaction having high activation energy and the chain displacement reaction having low activation energy. The VFD model with strong vibrational favored contribution can predict well the high temperature dissociation reaction, whereas the TCE model (a special case of VFD model) and QK model are better for the chain displacement reaction. Besides, the post-collision redistribution of internal energy should follow the principle of detailed balance, as small deviations may cause inequality between the translational and vibrational energy under final equilibrium state. The DSMC simulation results also show that the vibrational favor of chemical reactions has an obvious effect on the thermochemical coupling process. Particularly, because molecules having high vibrational energy are more easily to have chemical reactions, the decrease of the average vibrational energy of the gas will affect the subsequent chemical reactions.
YangChao, SunQuanhua. ANALYSIS OF DSMC REACTION MODELS FOR HIGH TEMPERATURE GAS SIMULATION 1)[J]. Acta Mechanica Sinica, 2018, 50(4): 722-733 https://doi.org/10.6052/0459-1879-18-056
An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions
.The Journal of Chemical Physics, 2015, 143(5): 054304
[35]
GimelsheinNE, GimelsheinSF, LevinDA.
Vibrational relaxation rates in the direct simulation Monte Carlo method
The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Models for direct Monte Carlo simulation of coupled vibration-dissociation
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
On the accuracy of DSMC modeling of rarefied flows with real gas effects
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
DSMC dissociation model based on two-temperature chemical rate constant
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Comparison of DSMC reaction models with QCT reaction rates for nitrogen
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
The QK model for gas-phase chemical reaction rates
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions
2016
Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations
2017
DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models
2017
A new approach for chemical reaction simulation of rarefied gas flow by DSMC method
2016
DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55th
2017
Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Hydrogen-oxygen detonation study by the DSMC method
1
2011
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation
1
2014
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method
... Clarendon Press, 1994樊菁. 稀薄气体动力学: 进展与应用. 力学进展, 2013, 43(2): 185-201(FanJing.Rarefied gas dynamics: Advances and applications.Advances In Mechanics, 2013, 43(2): 185-201 (in Chinese))HaasBL, BoydID.Models for direct Monte Carlo simulation of coupled vibration-dissociation.Physics of Fluids A: Fluid Dynamics, 1993, 5(2): 478-489BoydID, BoseD, CandlerGV.Monte Carlo modeling of nitric oxide formation based on quasi-classical trajectory calculations.Physics of Fluids, 1997, 9(4): 1162-1170BondarY, GimelsheinN, GimelsheinS, et al.On the accuracy of DSMC modeling of rarefied flows with real gas effects.AIP Conference Proceedings, 2005, 762(1): 607-613BondarYA, Ivanov MS. DSMC dissociation model based on two-temperature chemical rate constant. AIAA Paper, 2007-614, 2007WysongIJ, GimelsheinSF.Comparison of DSMC reaction models with QCT reaction rates for nitrogen.AIP Conference Proceedings, 2016, 1786(1): 050021BirdGA.The QK model for gas-phase chemical reaction rates.Physics of Fluids, 2011, 23(10): 106101BaikovBS, BayalinaDK, KustovaEV, et al.Inverse Laplace transform as a tool for calculation of state-specific cross sections of inelastic collisions.AIP Conference Proceedings, 2016, 1786(1): 090005LuoH, KulakhmetovM, AlexeenkoA.Ab initio state-specific N2+O dissociation and exchange modeling for molecular simulations.The Journal of Chemical Physics, 2017, 146(7): 074303SebastiãoIB, KulakhmetovM, AlexeenkoA.DSMC study of oxygen shockwaves based on high-fidelity vibrational relaxation and dissociation models.Physics of Fluids, 2017, 29(1): 017102RaminZ, Kamali-MoghadamR, ManiM.A new approach for chemical reaction simulation of rarefied gas flow by DSMC method.Computers & Fluids, 2016(140): 111-121SebastiaoIB, LuoH, KulakhmetovM, et al.DSMC implementation of compact state-specific N<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml229-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>+O dissociation and exchange models//55thAIAA Aerospace Sciences Meeting, 2017BoydID.Analysis of vibration-dissociation-recombination processes behind strong shock waves of nitrogen.Physics of Fluids A : Fluid Dynamics, 1992: 4(1): 178-185KimJG, BoydID.Monte Carlo simulation of nitrogen dissociation based on state-resolved cross sections.Physics of Fluids, 2014, 26(1): 012006WysongI, GimelsheinS, GimelsheinN, et al.Reaction cross sections for two direct simulation Monte Carlo models: Accuracy and sensitivity analysis.Physics of Fluids, 2012, 24(4): 042002WysongI, GimelsheinS, BondarY, et al.Comparison of direct simulation Monte Carlo chemistry and vibrational models applied to oxygen shock measurements.Physics of Fluids, 2014, 26(4): 043101ValentiniP, SchwartzentruberTE, BenderJD, et al.Direct molecular simulation of nitrogen dissociation based on an ab initio potential energy surface.Physics of Fluids, 2015, 27(8): 086102Bird GA. The DSMC Method.Create Space Independent Publishing Platform, 2013BirdGA.Chemical reactions in DSMC.AIP Conference Proceedings, 2011, 1333(1): 1195-1202BondarYA, MarutaK, IvanovMS.Hydrogen-oxygen detonation study by the DSMC method.AIP Conference Proceedings, 2011, 1333(1): 1209-1214YangC, SunQH.Investigation of spontaneous combustion of hydrogen-oxygen mixture using DSMC simulation.AIP Conference Proceedings, 2014, 1628(1): 1261-1267GimelsheinSF, GimelsheinNE, LevinDA, et al.On the use of chemical reaction rates with discrete internal energies in the direct simulation Monte Carlo method.Physics of Fluids, 2004, 16(7): 2442-2451SaxenaP, WilliamsFA.Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide.Combustion and Flame, 2006, 145(1): 316-323BenderJD, ValentiniP, NompelisI, et al.An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions.The Journal of Chemical Physics, 2015, 143(5): 054304GimelsheinNE, GimelsheinSF, LevinDA.Vibrational relaxation rates in the direct simulation Monte Carlo method.Physics of Fluids, 2002, 14(12): 4452-4455MaasU, WarnatzJ.Ignition processes in hydrogen oxygen mixtures.Combustion and Flame, 1988, 74(1): 53-69DoveJE, TeitelbaumH.The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr.Chemical Physics, 1974, 6(3): 431-444
The authors have declared that no competing interests exist. ...
Testing a small detailed chemical-kinetic mechanism for the combustion of hydrogen and carbon monoxide
An improved potential energy surface and multi-temperature quasiclassical trajectory calculations of <inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml230-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal">N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub><mml:mo>+</mml:mo><mml:msub><mml:mrow><mml:mi>N</mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula> dissociation reactions
2015
Vibrational relaxation rates in the direct simulation Monte Carlo method
The vibrational relaxation of H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml231-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>. I. Experimental measurements of the rate of relaxation by H<inline-formula><mml:math xmlns:mml="http://www.w3.org/1998/Math/MathML" id="Mml232-0459-1879-50-4-722"><mml:msub><mml:mrow><mml:mi mathvariant="normal"> </mml:mi></mml:mrow><mml:mrow><mml:mn>2</mml:mn></mml:mrow></mml:msub></mml:math></inline-formula>, He, Ne, Ar, and Kr



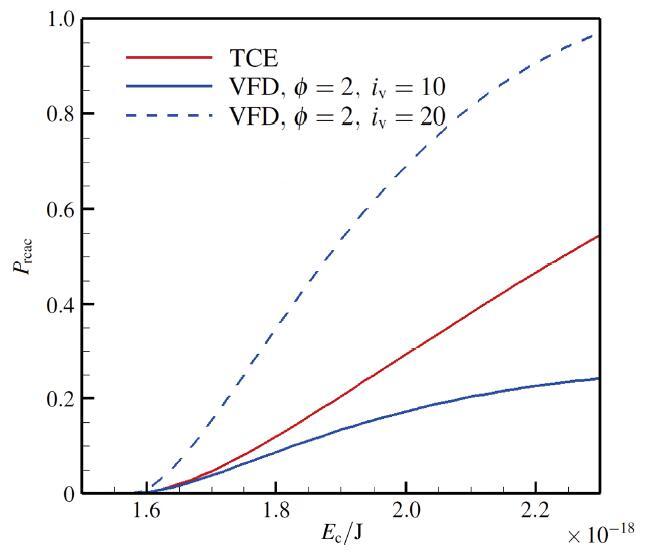
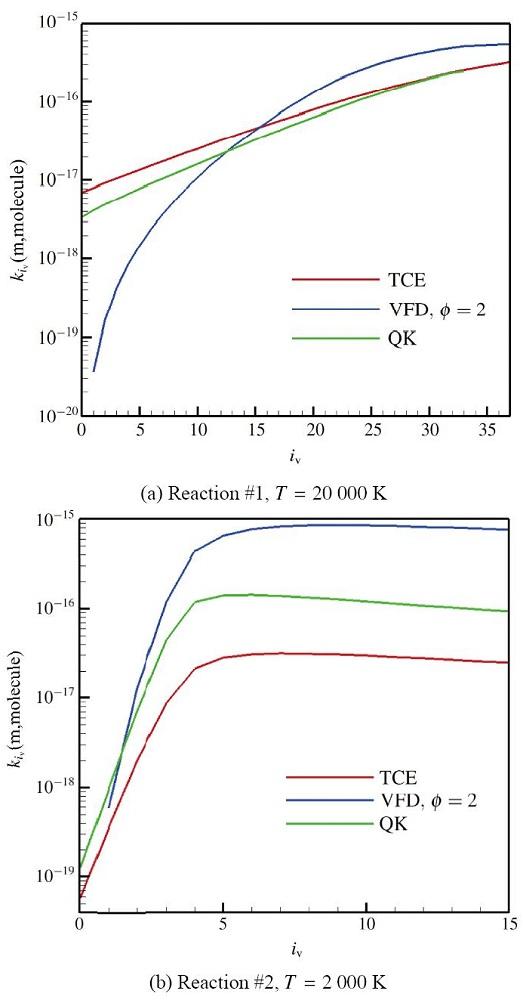
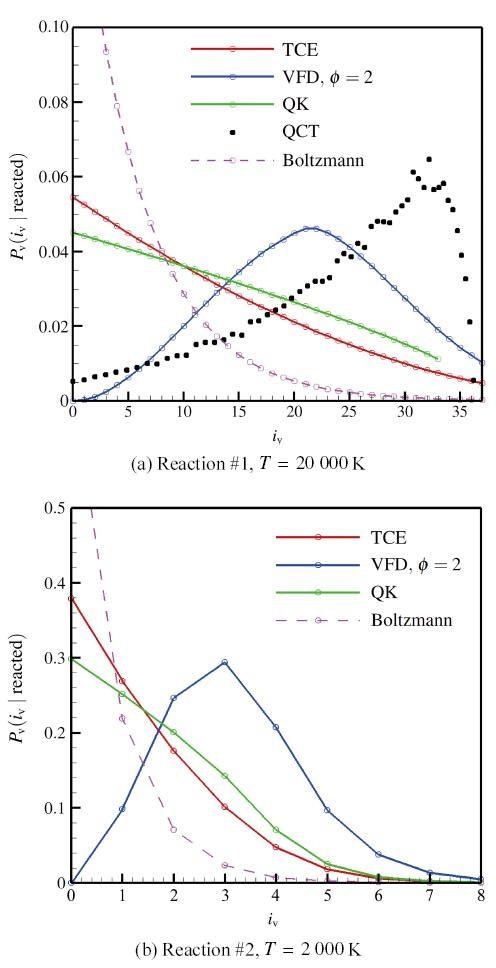

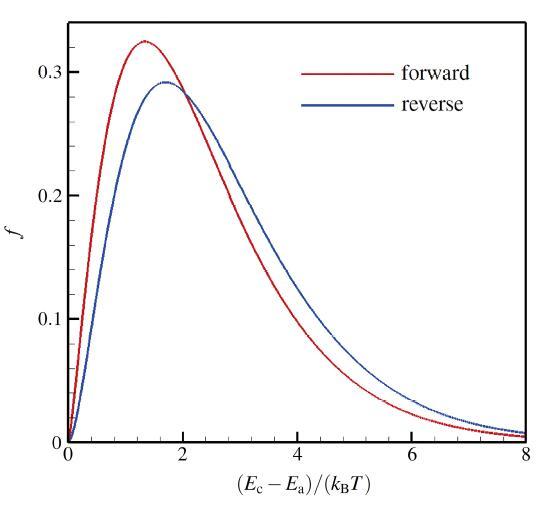
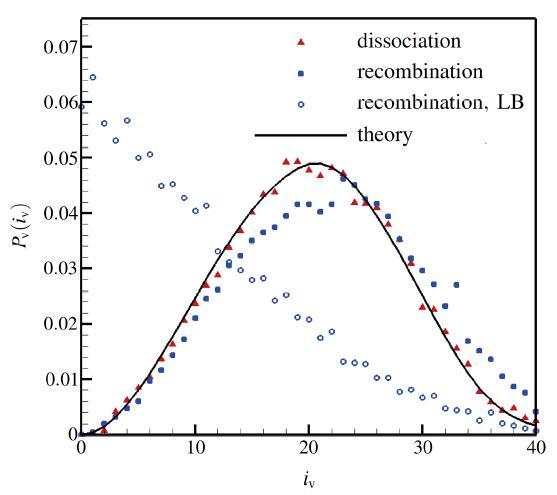
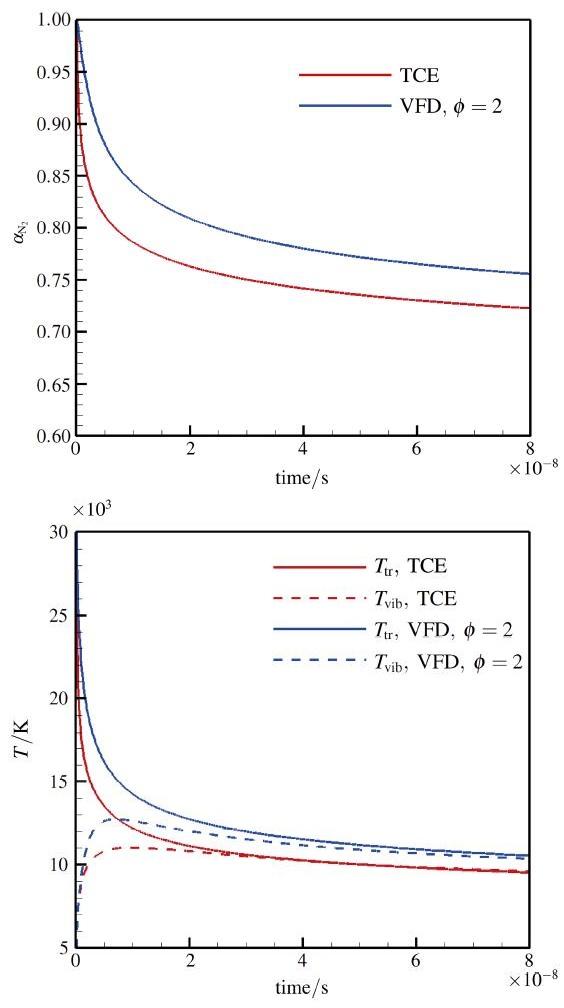
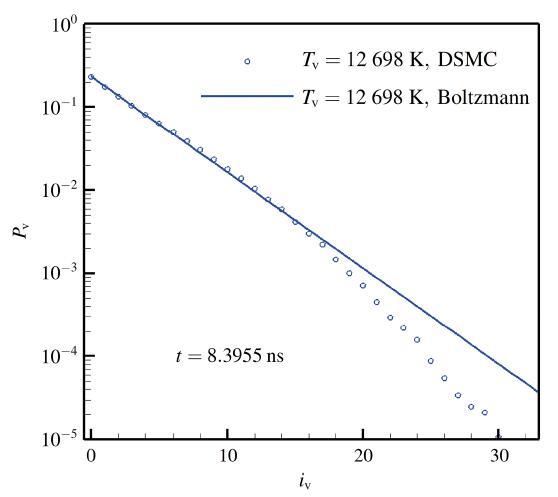

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
